Immune Dynamics of Tumor Neoantigen Recognition
As a member of the University of Maryland Greenebaum Comprehensive Cancer Center, the Perkins lab is interested in understanding how the innate immune system contributes to recognition of developing tumors and ultimately to tumor control and clearance.
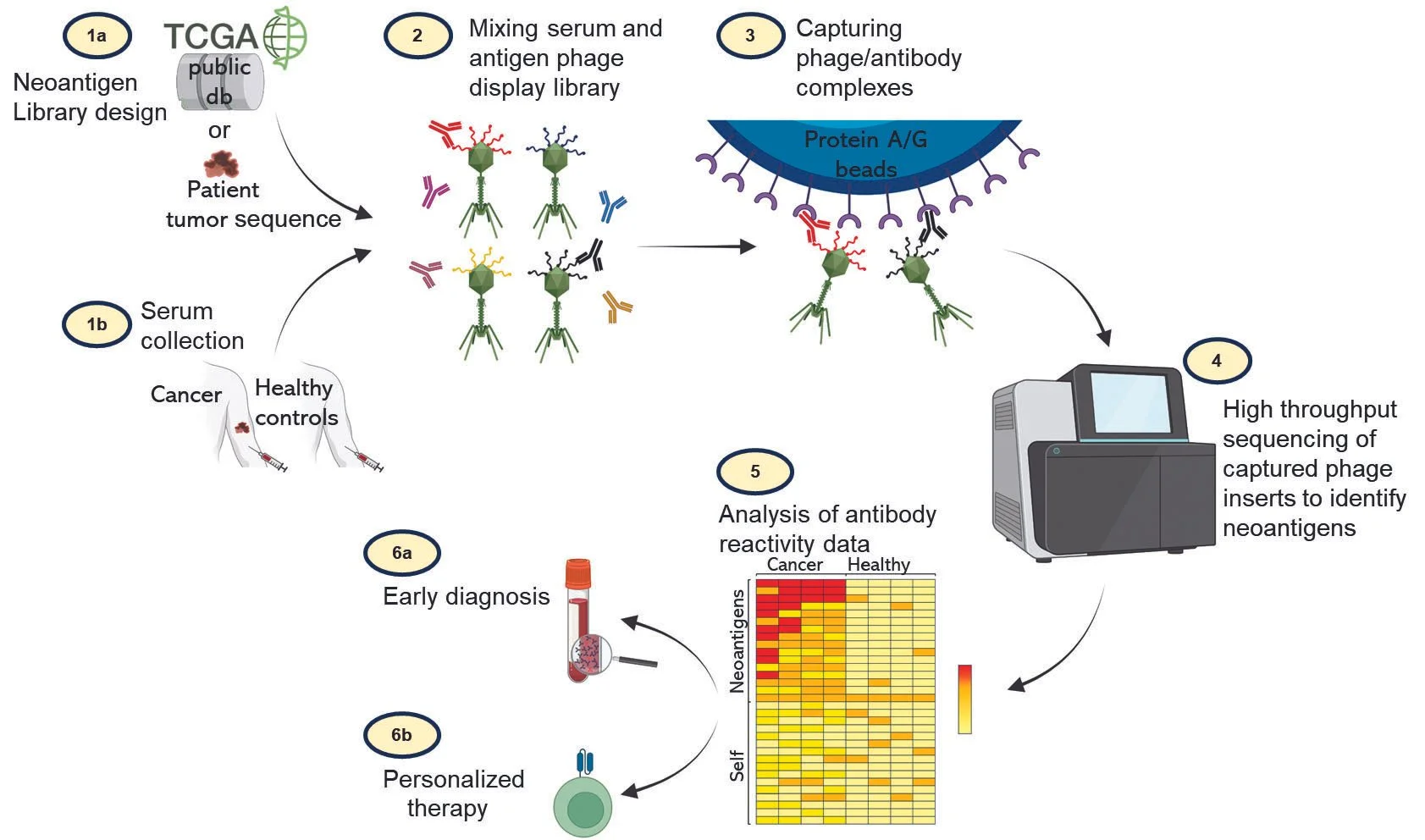
The cells in our immune system can recognize cancer as foreign because tumors express unique peptide antigens not expressed in healthy tissue. These tumor specific “neoantigens” arise due to acquired mutations in a tumor’s genomic DNA and can vary widely in number and sequence between types of cancers.
Understanding the neoantigen repertoire expressed by a given tumor allows us to address basic questions about the factors that influence immune recognition and to develop targeted immunotherapies such as CAR-T cells and cancer vaccines.
In collaboration with Dr. Thiagaraja Venkataraman at UMB we are applying a new systems immunology technology called PhIP-Seq to the simultaneous discovery and validation of tumor neoantigens. PhIP-Seq couples programmable phage display to next generation sequencing and allows us to profile circulating anti-tumor antibodies from small amounts of human or mouse sera. PhIP-Seq improves on the state of the art and is a high-throughput, rapid and scalable approach to determine immunogenic neoantigens and quantify the immune response to these antigens.
Cell Biologic Determinants of Innate Pattern Recognition Receptor Responses
The pattern recognition receptors of the innate immune system each have a complex cell biology which strongly influences the signals these receptors transmit and the transcriptional response programs they drive. We are interested in the many ways that the surrounding cell biological processes inform innate inflammatory responses to microbial signatures recognized by pattern recognition receptors.
Changes in cellular metabolic states, in particular lipid metabolism pathways, can qualitatively and quantitatively alter the inflammatory response to microbial triggers or to signatures of cancerous transformation, thereby contextualizing the innate response.
For example, we have previously found that the cell surface lipid receptor EP4 is a key regulator of TLR4s ability to signal through the endosomal TRIF pathway which does not affect cell surface MyD88 dependent signals (Perkins et. al. Nat Immuno 2018). EP4 sensing of eicosanoid lipid release licenses internalization and intracellular trafficking of TLR4 to late endosomes where TLR4 engages TRIF and drives a type I interferon response. This EP4 ‘lipid circuit’ allows the TLR4 driven inflammatory response to bacterial LPS to integrate information about a specific lipid metabolic pathway.
We have ongoing projects exploring the impacts of lipid metabolism on responses through the cGAS/STING sensor system located on the Endoplasmic Reticulum.
Transcriptomics and Epigenomics of Type I Interferons in Host Defense
The type I interferons (IFNs) are a family of cytokines which are the central pillar of our rapid immune response against all viral infections. In the absence of a functional IFN system, humans and animals rapidly succumb to otherwise harmless viral infections.
The IFNs function in viral infections as transcriptional regulators, increasing the expression of hundreds of Interferon Stimulated Genes or “ISGs”. These ISGs have direct antiviral functions and increase the resistance of each cell to an invading virus.
While critical to aiding our survival in the context of viral infections, type I interferons are abundantly expressed in other non-viral infectious and autoimmune diseases where they can paradoxically restrict innate and adaptive immune responses and profoundly shape the outcomes of disease.
Of particular interest to our group is the observation made by us and others that Type I IFN expression in the majority of pathogenic bacterial infections is harmful for the host and associated with a unique pattern of immune suppression.
It is difficult, if not impossible, to explain the immune suppressive actions of IFNs exclusively through ISGs, and we propose that the key to understanding the consequences of type I IFN expression in these non-viral diseases lies not in interferon stimulated genes but in interferon’s gene specific transcriptional suppression.
Transcriptional suppression remains a little explored transcriptomic space in the context of type I IFNs and the mechanistic molecules required are undefined.
Our ongoing work involves transcriptomics and epigenomics to define the ‘footprint’ of IFN transcriptional suppression through identifying all of the IFN suppressed genes specifically in the context of an ongoing inflammatory stimulus. We believe this work can ultimately lead to a deeper understanding of the deleterious consequences of IFNs in bacterial infections and the palliative effects of IFNs in autoimmune diseases such as multiple sclerosis.
Below are evolving descriptions of some of the ongoing avenues of investigation in the Perkins Lab.